


Contact Info
Adam P. Hitchcock
Canada Research Chair
in Materials Research
CLS-CCRS
B.I.M.R
McMaster University
Hamilton, ON
Canada L8S 4M1
V: +1 905 525-9140
x24729
F: +1 905 521-2773
E: aph@mcmaster.ca
U: unicorn.mcmaster.ca
__________
Research
Group
Opportunities
Publications
Links
_____________

|
|
An introduction for the non-specialist+
BIMR, McMaster University
Hamilton, ON Canada L8S 4M1
+ This document is intended to be an informal introduction of a tutorial nature, rather than an historical or comprehensive overview. Please see the articles referenced for more complete presentations of the technique and its many applications.
1. Introduction
X-ray absorption spectroscopy has advanced rapidly in the last
decades, particularly with synchrotron radiation based techniques [stohr
92]. The high brightness of the synchrotron X-ray source has allowed tremendous
improvements in spectral resolution and the development of X-ray microscopies
which apply X-ray absorption spectroscopy to heterogeneous materials at
high spatial resolution. Novel characterisation techniques are being developed
[ade 98] which allow both spectromicroscopy - imaging with spectral
sensitivity, and microspectroscopy - recording spectra from very
small spots. We choose to call these novel analytical tools X-ray spectromicroscopy,
to emphasize that it is the combination of spectroscopy and microscopy
that is the essence of these techniques. This article describes one of
these techniques, scanning transmission x-ray microscopy (STXM)
[ade 97, warwick 97, warwick 98], and illustrates its capabilities through
two selected applications in polymer analysis [ade 92, ade 93, ade 95,
smith 96]. Authoritative reviews of STXM have been published which stress
materials [ade 98, ade00] and biological [kirz 95] applications. 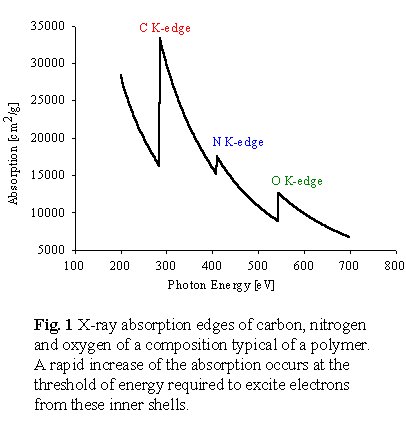
There are many problems in polymer physics and chemistry which require detailed chemical analysis at a sub-micron spatial scale - phase segregation; determination of the morphology and interface chemistry of blends and co-polymer systems; nano-patterned structures, self assembly; and many others. Traditional chemical spectroscopies used for polymer studies such as infrared and nuclear magnetic resonance can differentiate chemical species but they do not have the necessary sub-micron spatial resolution. Analytical transmission and scanning transmission electron microscopy have excellent spatial resolution and are very useful to visualise the structure, but electron microscopy typically does not have sufficient chemical sensitivity for quantitative chemical mapping beyond the elemental level. In many cases it is ambiguous whether structure observed by electron (or optical) microscopy arises from chemical differences or if it is simply caused by density/thickness variations or reflectivity changes (optical). In addition, electron microscopy of polymers is experimentally difficult on account of high rates of radiation damage by energetic electron beams [Egerton 87].
2. Near Edge X-ray Absorption Fine Structure
As an x-ray passes through matter, it is absorbed to an extent which
depends on the nature of the substance, the thickness of the sample, and
the density of the sample. The absorbed photons cause excitation of the
inner shell electrons of the atoms in the substance. These excited inner
shell (core) electrons can be promoted to unoccupied energy levels to
form a short lived excited state or they can be removed completely
to form an ionized state. Traditionally, X-ray absorption spectroscopy
was described in terms of absorption edges which are the onsets
of inner-shell ionization (Figure 1). There is an absorption edge
associated with each inner shell energy level of an atom, such that all
elements have an X-ray absorption edge in the soft X-ray energy range
(100-1200 eV). The amount of a particular element can be determined quantitatively
from the difference in the x-ray absorption just above and just below
its absorption edge. 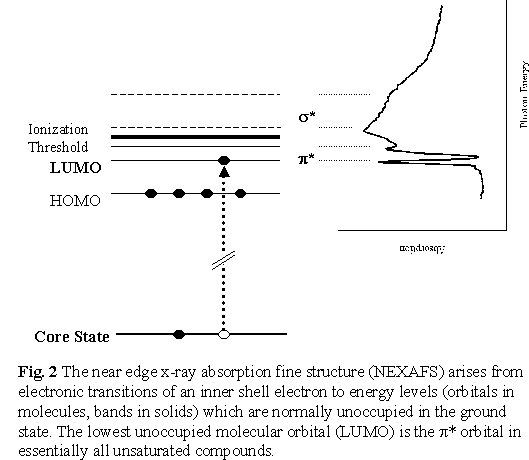
While this traditional aspect of X-ray absorption provides the basis for
elemental analysis, the modern technique as practised at synchrotron facilities
is much more powerful. It can identify and quantitate the chemical structure
of the element from the fine scale details of the absorption spectrum
that occur at each edge, the so-called near-edge x-ray absorption fine
structure or NEXAFS [Stohr 92]. These features, which can be as much
as 10 times stronger than the absorption edge jump, correspond to electronic
excited states in which an inner-shell electron has been excited to unfilled
molecular orbitals or conduction bands. As the x-ray energy is increased
throughout an absorption edge, first there is structure associated with
excitation to the lowest unoccupied molecular orbital, which is a p
* orbital for unsaturated molecules (double or triple bonds), followed
by structures associated with higher energy unoccupied molecular orbitals,
typically of s * character associated with
saturated (single) chemical bonds, and then direct inner-shell ionization
(Figure 2).
The unoccupied electronic structure and thus the inner-shell excited
states are determined by the geometric and electronic (bonding) structure
of the sample. NEXAFS spectra differ significantly even for rather similar
molecular structures, as shown in Figure 3 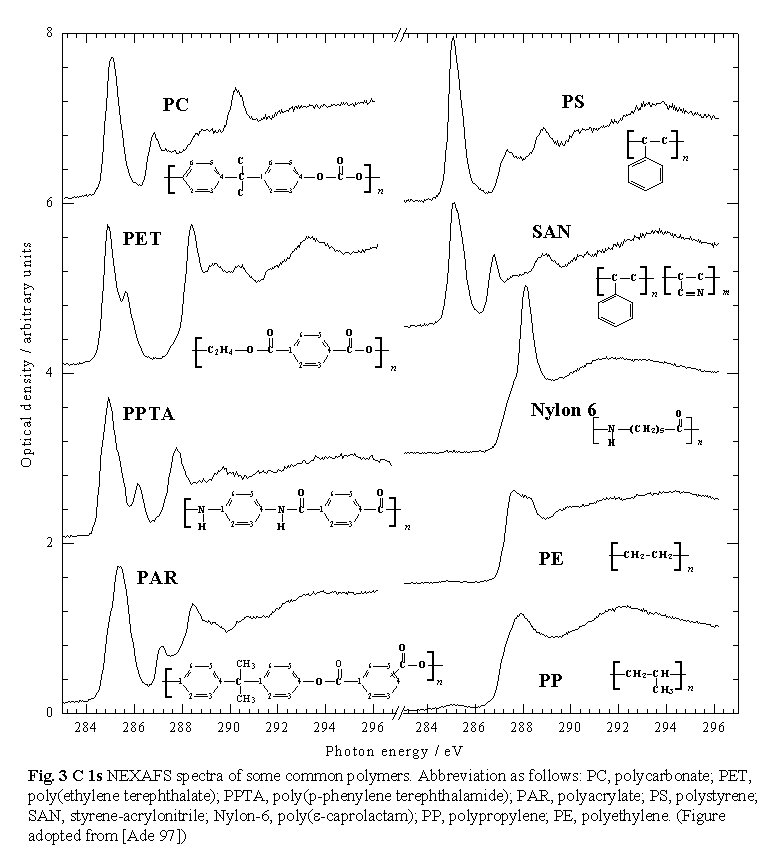 for
some common polymers [Ade 97b]. This means that the NEXAFS spectrum of
each polymer can be used as a fingerprint. In many cases, enough
is known about how chemical structure and X-ray absorption spectral features
are related to allow one to identify unknown species from measured NEXAFS
spectra. Individual spectral features, particularly the low energy p
* features, are often sufficient for qualitative identification in reasonably
well characterized systems, and they can serve as useful energies for
selective chemical contrast in X-ray microscopy.
for
some common polymers [Ade 97b]. This means that the NEXAFS spectrum of
each polymer can be used as a fingerprint. In many cases, enough
is known about how chemical structure and X-ray absorption spectral features
are related to allow one to identify unknown species from measured NEXAFS
spectra. Individual spectral features, particularly the low energy p
* features, are often sufficient for qualitative identification in reasonably
well characterized systems, and they can serve as useful energies for
selective chemical contrast in X-ray microscopy.
There is characteristic NEXAFS structure at the absorption edge of each element in a sample. Thus combined studies of C 1s, N 1s and O 1s NEXAFS is a very powerful tool in polymer analysis. Finally, comparisons with NEXAFS spectra of pure standards provides a means to derive quantitative composition maps of the components of a complex material from a series of X-ray microscopy images.
3. Scanning Transmission X-ray Microscope (STXM): Simple Description
and Operation
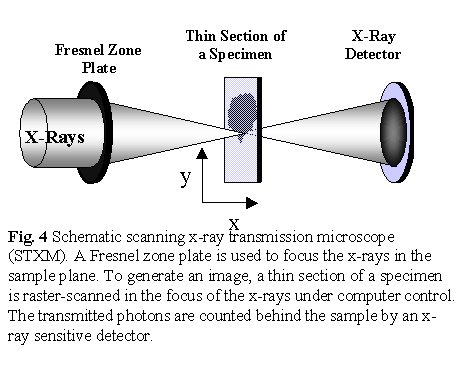
A simplified view of STXM is shown in Figure 4. A Fresnel zone
plate focuses mono-energetic X-rays provided by a suitable monochromator
beamline mounted on a suitable, bright synchrotron source. The focal point
is typically 50 nm in diameter over a 3-10 mm
waist. An image is generated by monitoring the X-ray signal transmitted
through a thin section of a specimen as it is raster-scanned at the focus
of the x-rays. Micro-spectra are measured by holding the beam at the spot
of interest on the sample while the photon energy is scanned. The sample
in a STXM can be mounted in air (with sufficiently short X-ray path length),
in He at atmospheric pressure, or even sandwiched between two X-ray transparent
silicon nitride windows. The latter approach is used to study wet samples
such as hydrated polymers [Mitchell99] or biological material [Kirz95].
The STXM at the Advanced Light Source (ALS) which we use can record images with less than 50 nm spatial resolution, and NEXAFS spectra from 150 - 1250 eV on stxm701 (210 - 650 eV on stxm5.3.2) with an energy resolution of about 100 meV [warwick 97, warwick 98]. Samples must be partly transparent at the X-ray energy of interest, which means a sample thickness of 50 to 300 nm for carbon studies, ranging up to 1 - 2 microns thick for higher energy edges or low density samples such as hydrated polymer gels or biological samples. The sample preparation challenges are similar to those encountered for analytical transmission electron microscopy. Microtomed sections about 100 nm thick which are mounted on 3 mm metal grids are a common form of sample. We use silicon wafers with X-ray transparent, 100 nm thick silicon nitride windows (of up to 4 mm lateral size) to study samples in water. One can even do N 1s spectroscopy through these windows. A strength of STXM relative to electron microscopy is that radiation damage is two orders of magnitude less than in electron beam imaging techniques [rightor 97], making this technique ideally suited for radiation sensitive polymers.
The transmitted X-ray signal is measured by single photon counting using counting periods (dwell times) of 0.2 - 0.5 ms per pixel for imaging. A 300x300 pixel image takes about 30 seconds to acquire. Typically, many problems can be solved by measuring a small number of images at selected, chemically sensitive images. In cases where one needs a very detailed chemical analysis of a specific region it is useful to take a systematic series of images at a fine mesh of energies around the absorption edge of interest. These image sequences - which might involve one hundred 100x200 pixel images, or about 8 Mb of raw data - can be processed off-line in a variety of ways for both qualitative species identification and to derive quantitative composition maps. Examples of these approaches are shown below.
For quantitative analysis, the transmitted signal is converted to an optical density (OD) according to
OD = ln(Io/I)
where for a given X-ray energy I0 is the incident x-ray flux, I is the transmitted flux through the sample, and ln is the natural logarithm. The OD is related to the sample properties by
OD = µ(E).r .t
where m(E) is the mass absorption coefficient at X-ray energy E, r is the density and t is the sample thickness. The mass absorption coefficient is derived from measurements of the NEXAFS spectra of the pure material. Practically, spectra are obtained by first recording an energy scan I from the spot of interest and subsequently the incident flux Io measured with the same detector and optical path but with the sample out of the beam. Typically, this takes a few minutes.
4. Example 1: Qualitative polymer analysis
This is part of a study [Hitchcock 01] of reinforcing filler particles
which are used in moulded compressed polyurethane foams in the automotive
and furnishing industries to achieve higher hardness and load bearing
capability. STXM was been used to study two types of copolymer polyol
(CPP) fillers hard polymer particles dispersed in a polyether polyol
- present in a toluene di-isocyanate-based polyurethane. One CPP is a
copolymer styrene and acrylonitrile (SAN). The other CPP is an aromatic-carbamate
rich poly-isocyanate poly-addition product
(PIPA), derived from methylene diisocyanate. Both particles are chemically
indistinguishable by transmission electron microscopy (Figure 5)
although the size and spatial distribution are clearly revealed.
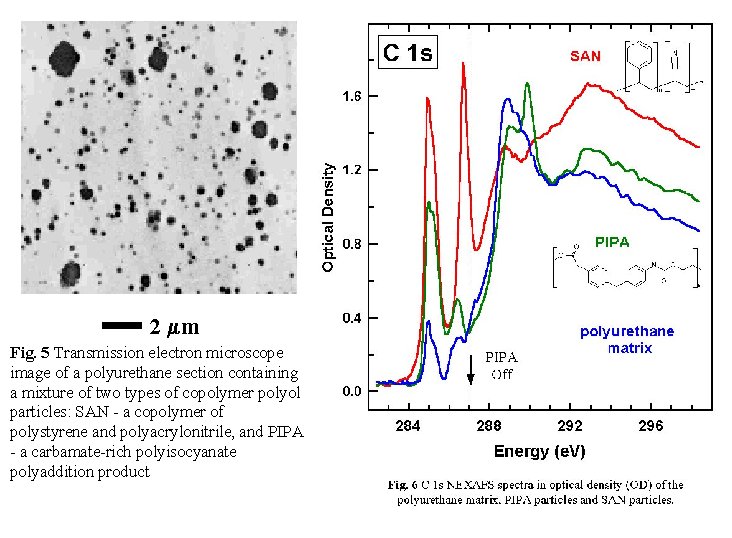
Chemical selective imaging in the C 1s NEXAFS (Figure 6) identifies
the SAN and PIPA particles unambiguously. Since the spectra of both SAN
and PIPA absorb strongly at 285.0 eV associated with the phenyl groups
of the aromatic filler particles, images at 285.0 eV show both types of
particles and thus the 285 eV image (Figure 7a) is identical to
the electron microscopy image. Only SAN has a strong absorption
at 286.7 eV, associated with the acrylonitrile component (AN). Thus imaging
at 287 eV, on the higher energy side of this peak where the optical density
of the PIPA particles and the matrix is the same and low compared to the
SAN particles, gives an image in which only SAN particles are in high
contrast (Figure 7b). After subtracting the 287 eV from the 285
eV image, the PIPA particles can be identified without difficulty (Figure
7c). This example dramatically demonstrates that NEXAFS microscopy
can be a quick and reliable means to differentiate chemical species at
a sub-micron spatial scale. Further analysis of this material has lead
to full quantification of the CPP particle compositions, and detailed
size distribution analyses for each type of particle. Neither of these
aspects would have been achievable by analytical electron microscopy.

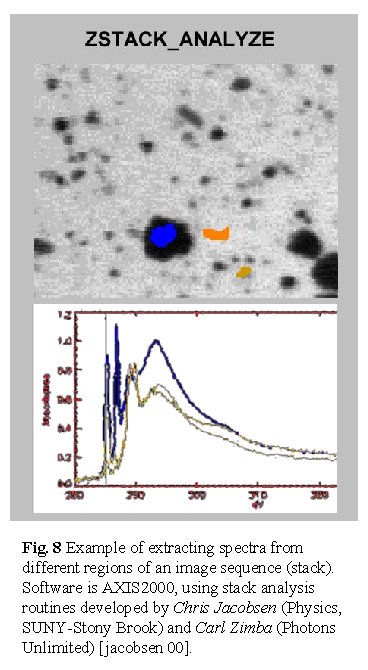
5. Image processing and advanced results
Recording sequences of images with short dwell time and fine energy spacing
can be very useful [jacobsen 00]. For instance, post-processing of the
images series can be used to extract spectra at any subset of pixels in
the sampled region, long after the measurement is finished. This approach
can provide serendipitous discoveries. An example of this for the PIPA-SAN
polyurethane filler particle system is shown in Figure 8. Relative
to multiple single point measurements, image sequence procedures also
minimize radiation damage. At the ASL point spectra cannot be acquired
without exposing the sample to ~200 msec per energy point, or say, 20
seconds for a 100 point energy scan. In contrast, image sequences which
can provide statistical precision of 1-2% in a 0.2x0.2 m
m region can be acquired with dwell times per pixel of 0.5 msec, or 50
msec total for the full 100-point energy spectrum. Of course damage is
not restricted solely to the point sampled but even so, there is a reduction
in dose of several orders of magnitude relative to point scan mode.
Another application of image sequences is quantitative analysis.
The absorption signal from a given column of the sample is related to
the amount of each component, the x-ray absorption response of each component,
and the overall thickness and density of the material in the column. Since
NEXAFS spectra for pure materials can be obtained with accurate mass absorption
scales, STXM can be used for quantitative analysis at sub-micron ranges
[Koprinarov 00]. Quantitative composition maps can be derived by two different
approaches singular value decomposition (SVD) [Koprinarov 00] and
image sequence fits [Kneedler 00]. A brief description of each
follows.
Singular value decomposition
The optical density is determined by the formula OD = µ.r .t, where µ is the energy-dependent mass absorption coefficient (cm2/g), r is the density (g/cm3), and t is the sample thickness (cm). If the mass absorption coefficients of each components are known at the energies sampled, the problem can be converted to a simple superposition and linear algebra can be used to convert sets of images into equivalent thickness (rt(x,y)) or composition maps. In principle, the decomposition problem can be expressed as a matrix equation Ax=d, where x is a vector describing the unknown distribution of each component (rt), d are the measured images (converted to OD scale), and A is the matrix of the mass absorption coefficients (µ) obtained from reference spectra for each component, converted to mass absorption scale. The advantage of the singular value decomposition procedure is that, once the absorption coefficients for a set of materials and energies are known, one can calculate a priori the matrix which will optimally invert an over-determined sampling of energies into the best possible component maps. It can be shown that the SVD solution is the least squares solution [strang 88, press 92].
Image sequence fits
Image sequence fitting [hitchcock 02] computes a least squares fit of the intensity at each pixel(j,k) to a linear combination of reference spectra for each component (converted to mass absorption scale). An energy-independent (constant) term (ao) is also usually included to accommodate unexpected backgrounds:
_________________
6. Example 2; Quantitative chemical mapping at sub-micron resolution
The second example illustrates an application of image sequence analysis
[Croll00]. Polymer capsules and particles with tailored walls,
and core-shell structures are attractive for a wide number of applications,
including separation science applications, adhesives, coating, chemical
delivery, etc. [Li99]. In order to optimise for a particular application,
e.g. designing the wall properties for controlled release of a specific
chemical, one needs to perform accurate quantitative analysis of the chemical
structure of the wall at high spatial resolution. The polyurea capsules
are made by dispersing a mixture of aromatic isocyanate and xylene in
an aqueous solution of a polyamine. Interfacial polymerisation reactions
of both the amine and water with the isocyanate occur at the surface of
the dispersed hydrophobic, organic droplets. Competition among amine-isocyanate
and water-isocyanate reactions is controlled by differences in kinetics
and diffusion rates. This can lead to chemically structured capsule walls.
The amine-isocyanate reaction forms an asymmetric (aromatic-aliphatic)
urea whereas the water-isocyanate forms a symmetric di-aromatic urea.

We have used quantitative NEXAFS microscopy to visualize the gradient
of chemical composition across a capsule wall formed in this manner. Figure
9 presents component maps derived from image sequences using singular
value decomposition and model spectra taken from small molecule analogues.
The vertical scale of these maps represents the thickness in nm of that
component. The profiles across the capsule wall in the region marked on
the maps, show that there is an internal chemical structure to the capsule
wall, and that this structure reflects the expected changes arising from
the synthetic methodology. In particular, the symmetric di-aromatic urea
from the water-isocyanate reaction is a narrower band at the outer region
of the wall, whereas the asymmetric urea arising from the amine-isocyanate
reaction is thicker and extends all the way to the inner side of the capsule
wall. Please note that the total wall thickness is only 500 nm and the
differences in chemical composition are observed with better than 100
nm spatial resolution. This precision of chemical quantification at high
spatial resolution demonstrates that STXM is a powerful analytical tool
which can provide assistance in the development of structured polymeric
systems which cannot be provided by any other technique.
Outlook
There are only a few STXM microscopes in the world. The instrumentation was originally developed by Kirz and collaborators (SUNY-Stony Brook) and the Kirz and Jacobsen groups operate three STXM at the X1 undulator beamline at the National Synchrotron Light Source in Brookhaven. At the Advanced Light Source, there are two operating STXM microscopes - one on beamline 7.0 (to be moved to beamline 11.0 in fall 2002); the other on bending magnet beamline 5.3.2 which is dedicated to polymer and soft materials analysis. A third centre for STXM is the Kings College instrument at beamline 5U2 in Daresbury, England. Other STXM instruments are under construction at BESSY II in Berlin, at the Swiss Light Source, at PLS, the Korean synchrotron, and at the Canadian Light Source.
If you are interested in analytical studies of polymers or other materials using STXM or other X-ray microscopy techniques, please contact
BIMR
McMaster University
Hamilton, ON L8S 4M1 Canada
Tel: 905 525-9140 ext 24749
E-mail: aph@mcmaster.ca
We thank Harald Ade for sharing beam time, results and a common perspective. The contributions of many people who helped develop, optimise and maintain the ALS STXM have been critical - we note in particular the contributions of Tony Warwick and George Meigs. The ALS BL 7.0 STXM was developed by T. Warwick (ALS), B.P. Tonner and collaborators, with support from the U.S. DOE under contract DE-AC03-76SF00098. The research described in this article is funded by strategic and research grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
References
[ade 92] H. Ade, X. Zhang, S. Cameron, C. Costello, J. Kirz, and S. Williams, Science, 258, 972 (1992)
[ade 93] H. Ade and B. Hsiao, Science, 262, 1427 (1993)
[Ade 95] H. Ade, A. Smith, S. Cameron, R. Cieslinski, C. Costello, B. Hsiao, G.E. Mitchell, and E.G. Rightor, Polymer, 36, 1843 (1995).
[ade 97] H. Ade, A.P. Smith, H. Zhang, B. Winn, J. Kirz, E. Rightor and A.P. Hitchcock, J. Electron Spectrosc. 84, 53 (1997).
[ade 98] H. Ade, in Experimental Methods in the Physical Sciences, Vol. 32, pp. 225, J.A.R. Samson and D.L. Ederer Ed., Academic Press, 1998.
[ade 00] H. Ade and S.G. Urquhart, Chemical Applications of Synchrotron Radiation ed. TK Sham, (World Scientific, 2000, in press)
[Croll 00] L.M. Croll, I. Koprinarov, A.P. Hitchcock, W.H. Li, and H. Stöver, in preparation.
[Egerton 87] R. E. Egerton, P. A. Crozier, P. Rice, Ultramicroscopy, 23, 305 (1987).
[hitchcock 01] A. P. Hitchcock, I.Koprinarov, T. Tyliszczak, E. G. Rightor, G. E. Mitchell, M. T. Dineen, W. Lidy, R. D. Priester, S. G. Urquhart, A. P. Smith, H. Ade, Ultramicroscopy 88, 33 (2001).
[hitchcock02] A.P. Hitchcock, I. Koprinarov, L.M. Croll, H. Stover and E.M. Kneedler, in preparation
[jacobsen 00] C. Jacobsen, S. Wirick, G. Flynn, and C. Zimba, J. Microscopy 197, 173 (2000).
[Kirz96] J. Kirz, C. Jacobsen and M. Howells, Quarterly Review of Biophyisics, 33 , 33 (1995).
[Koprinarov 00] I. Koprinarov, A.P. Hitchcock, K. Dalnoki-Varess, C. Murray, J. Dutcher, and H. Ade, unpublished.
[Li99] W.-H. Li and H.D:H Stöver, J. Polym. Sci., Polym. Chem. 37, 2295 (1999).
[mitchell99] G.E. Mitchell et al, ALS Compendium of Abstracts, 1999
[press 92] W.H. Press, et al. Numerical Recipes in C: The Art of Scientific Computing. Cambridge: Cambridge University Press, 1992.
[rightor 97] E.G. Rightor, A.P. Hitchcock, H. Ade, R.D. Leapman, S.G. Urquhart, A.P. Smith, G. Mitchell, D. Fischer, H.J. Shin and T. Warwick, J. Phys. Chem. B 101, 1950 (1997).
[Smith 96] A. P. Smith and H. Ade, Appl. Phys. Lett. 69, 3833 (1996)
[Stohr 92] J. Stöhr, NEXAFS Spectroscopy, Springer-Verlag, Berlin, 1992.
[strang88] G. Strang, Linear Algebra and Its Applications. San Diego: Harcourt Brace Jovanovich, 1988
[warwick 97] T. Warwick, H. Padmore, H. Ade, A.P. Hitchcock, E.G. Rightor and B. Tonner, J. Electron Spectrosc. 84, 85 (1997).
[warwick 98] T. Warwick, K. Franck, J.B. Kortwight, G.
Meigs, M. Moronne, S. Myneni, E. Rotenberg, S. Seal, W.F. Steele, H. Ade,
A. Garcia, S. Cerasari, J. Denlinger, S. Hayakawa, A.P. Hitchcock, T.
Tyliszczak, E.G. Rightor, H.-J. Shin and B. Tonner, Rev. Sci. Inst.
69, 2964 (1998).
|
|
© 2002 A.P. Hitchcock / McMaster University
- All Rights Reserved
web site by Christopher Amis. Last updated on 04/23/2002 (aph)